氯虫苯甲酰胺(chlorantraniliprole)是由美国杜邦公司于2000年研究开发的一种对鳞翅目夜蛾科、螟蛾科、蛀果蛾科等均有很好的控制效果的第一代鱼尼丁受体抑制剂类农用杀虫剂。
氯虫苯甲酰胺具有新颖的作用方式,通过使昆虫鱼尼丁受体的活化达到防治害虫的目的,因其具有高效低毒、环境友好、广谱等特性而被广泛使用。氯虫苯甲酰胺2021年以17.04亿美元销售额的领先优势,确立并夯实了它在全球杀虫剂市场的领军地位,如今其生产专利已到期,国内多家公司布局氯虫苯甲酰胺原药及其中间体产业链,全部达产后预计可形成5.7万t原药与1.5万t中间体产能,具有广阔的市场空间及发展前景。
目前,文献和专利所报告的关于氯虫苯甲酰胺的合成方法众多,这些方法大多都是以2,3-二氯吡啶为原料。最初杜邦公司是以N,N-二甲基氨基磺酰基吡唑为原料,在强碱低温条件下先进行溴化,再与2,3-二氯吡啶进行偶联,然后引入羧基得关键中间体3-溴-1-(3-氯-2-吡啶基)-1H-吡唑-5-甲酸。
随后对该工艺进行改进,经缩合、加成环和、溴代、氧化、水解等步骤得到关键中间体3-溴-1-(3-氯-2-吡啶基)-1H-吡唑-5-甲酸,最后在甲磺酰氯作用下与2-氨基-5-氯-N,3-二甲基苯甲酰胺反应得到目标产物(合成路线见图1)。该方法步骤繁琐,工艺路线复杂,设备投资较大,同时反应过程中使用高沸点溶剂及甲基磺酰氯等剧毒管制药品,反应工艺危险性也较高。
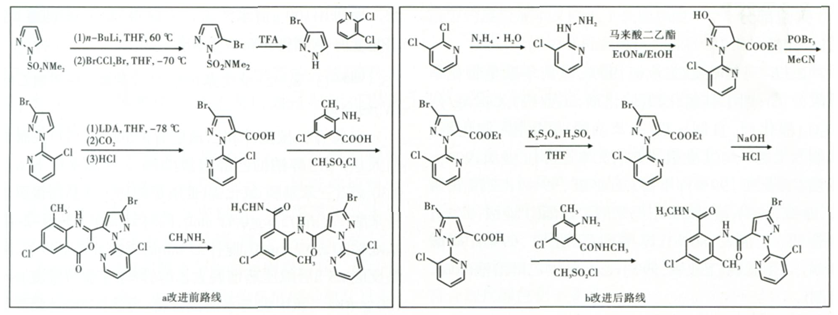
图1 氯虫苯甲酰胺的常用合成路线1
另外,冯显国以2,3-二氯吡啶为起始原料,经缩合、加成环和、溴化、水解得关键中间体3-溴-1-(3-氯-2-吡啶基)-4,5-二氢-1H-吡唑-5-甲酸,中间体3-溴-1-(3-氯-2-吡啶基)-4,5-二氢-1H-吡唑-5-甲酸经二氯亚砜酰化氧化后,与2-氨基-5-氯-N,3-二甲基苯甲酰胺反应得到目标产物(合成路线见图2)。
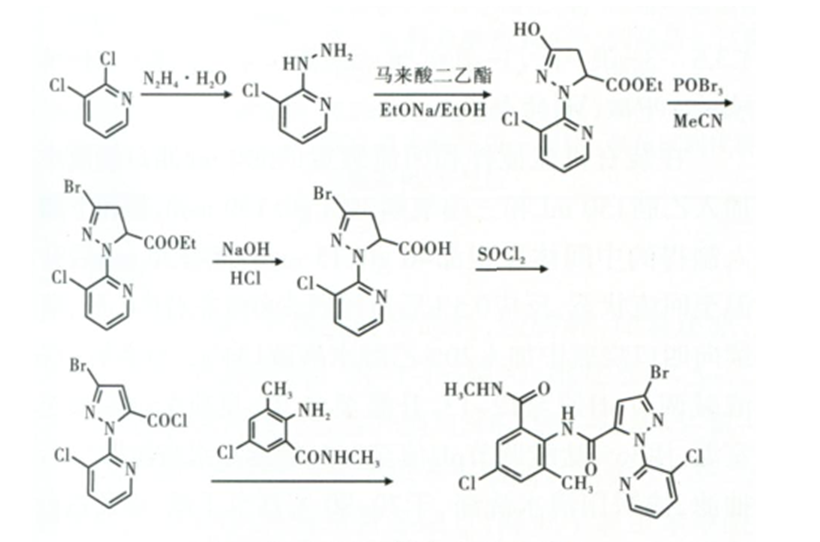
图2 氯虫苯甲酰胺合成路线2
在此基础上,本文以2,3,6-三氯吡啶(I)为原料,在四丁基氯化铵作用下与水合肼反应得到3,6-二氯-2-肼基吡啶(II),然后在Cu-Ag/γ-Al2O3的催化作用下,加氢还原反应得到3-氯-2-肼基吡啶(Ⅲ),中间体Ⅲ再与马来酸二乙酯在1%双(三苯基膦)氯化镍的催化作用下制得2-(3-氯-2-吡啶基)-5-吡唑烷酮-3-甲酸乙酯(Ⅳ),中间体Ⅳ经“一锅法”溴代一水解得到3-溴一1-(3-氯-2-吡啶基)-4,5-二氢-1H-吡唑-5-甲酸(Ⅵ),再由(Ⅵ)先后与SOCl2和2-氨基-5-氯-N,3-二甲基苯甲酰胺(Ⅷ)反应制得氯虫苯甲酰胺(Ⅸ),合成路线见图3。
与文献报道的方法相比,本文所开发的方法原料易得、价格较为低廉,同时使用一锅法合成3-溴-1-(3-氯吡啶-2-基)-4,5-二氢-1H-吡唑-5-甲酸,既避免了低温反应又降低了对设备的要求,另外本方法还具有经济性高、副反应少、反应条件温和、操作简便安全和对环境污染小等优点。
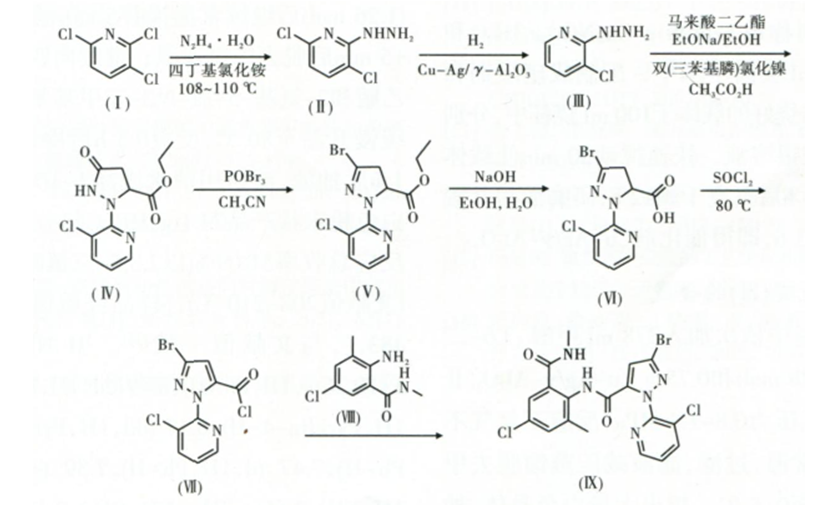
图3 本文氯虫本家酰胺的合成路线
1. 试剂
2,3,6-三氯吡啶(工业级,99%,重庆华歌生物化学有限公司);四丁基氯化铵(催化剂,工业级);Cu-Ag/γ-Al2O3(催化剂,自制);双(三苯基膦)氯化镍(催化剂,自制);二氯甲烷(工业级,99%);无水乙醇(工业级,99%);乙腈(工业级,99%);甲醇(工业级,99%);三溴氧磷(工业级,99%);液碱(30%);马来酸二乙酯(工业级,97%);水合肼(工业级,80%);冰醋酸(工业级,99%);盐酸(36%);二氯亚砜(工业级,99%);乙醇钠的乙醇溶液(20%,自制)。
2. 仪器及检测条件
仪器:岛津高效液相色谱仪[LC-16,岛津企业管理(中国)有限公司南京分公司]。
检测条件:C18硅胶反相柱(4.6 mmx150 mm);流动相为乙腈:水:醋酸=1:1:0.01;柱温38℃;检测波长254 nm;流速1 mL/min。
3. 中间体及目标化合物的合成
(1)3,6-二氯-2-肼基吡啶(Ⅱ)的合成
在装有回流装置的500 mL四口烧瓶中依次加入2,3,6-三氯吡啶60 g(0.33 mol)和80%水合肼溶液96.6 g(1.54 mol),2者混合后在常温搅拌下加入四丁基氯化铵1.8 g(0.006 mol),然后升温至108~110℃,回流反应6h后冷却至0~10℃,析出大量白色固体,抽滤,滤饼用清水洗涤,干燥得57.50 g中间体Ⅱ,HPLC归一含量99.2%,收率97.4%。
(2)催化剂Cu-Ag/γ-Al2O3的制备
载体预处理:取一定量的γ-Al2O3在马弗炉中500℃焙烧3 h。
催化剂制备:分别称取1.4496 g Cu(NO3)2·3H2O和1.0189 g AgNO3溶于3 mL去离子水中,配制成硝酸铜和硝酸银溶液;称取5.0 g焙烧好的载体于100 mL烧杯中,分别加入2 mL硝酸铜和硝酸银溶液,快速搅动20 min使载体呈黏糊状,室温下静置4h后真空干燥12 h,研磨成粉并置于马弗炉中450℃焙烧3 h,即得催化剂Cu-Ag/γ-Al2O3。
(3)3-氯-2-肼基吡啶(Ⅲ)的合成
在500 mL的加氢釜中依次加入278 mL甲醇、3,6-二氯-2-肼基吡啶50 g(0.28 mol)和0.75 g Cu-Ag/γ-Al2O3(Ⅱ质量的1.5%),控制氢气压力0.8-1.4 MPa,反应至氢气不再参与反应后冷却至常温,过滤,滤液减压蒸馏脱去甲醇后加入180 g水降温至0~5℃,析出大量白色晶体,抽滤,滤饼用100 g清水洗涤,干燥得32.54 g中间体Ⅲ,HPLC归一含量99.1%,收率80%。
(4)2-(3-氯-2-吡啶基)-5-吡唑烷酮-3-甲酸乙酯(Ⅳ)的合成
在装有机械搅拌和回流装置的500 mL四口烧瓶中加入147 g乙醇钠的乙醇溶液(20%),在冰水浴下加入0.30 g双(三苯基膦)氯化镍(Ⅲ质量的1%),然后缓慢滴加马来酸二乙酯75.6 g(0.43 mol),同时分批加入3-氯-2-肼基吡啶30 g(0.21 mol),搅拌25 min后缓慢升温至45℃,保温反应0.5 h后减压蒸馏脱去乙醇;降至室温后缓慢加入150 g 40%二氯甲烷与水的混合物,再滴加70%冰醋酸水溶液,调节pH值至5~7后继续搅拌1 h,静置1 h后分出油相,油相常压蒸馏除去二氯甲烷,得红棕色油状粗品46.96g,HPLC归一含量90%,收率75%。
(5)3-溴-1-(3-氯吡啶-2-基)-4,5-二氢-1H-吡唑-5-甲酸(Ⅵ)的合成
在装有机械搅拌和回流装置的500 mL四口烧瓶中加入乙腈150 mL和三溴氧磷39.8 g(0.139 mol),搅拌下加入制得的中间体Ⅳ粗品46 g(0.15 mol).搅拌20 min后升温至回流状态,反应0.5 h后负压脱去80%左右的乙腈:继续向四口烧瓶中加入20%乙醇水溶液134 g,再滴加30%液碱调节pH值至12~13,升温至45℃,反应0.5 h后降至室温,用36%盐酸调节pH值至2~3,继续冰水浴搅拌2 h后抽滤,滤饼用清水洗涤,于70~80℃真空干燥,得黄白色粉末50.93 g,HPLC归一含量95%,收率93%。
(6)氯虫苯甲酰胺(Ⅸ)的合成
在装有机械搅拌和回流装置的500 mL四口烧瓶中加入3-溴-1-(3-氯吡啶-2-基)-4,5-二氢-1H-吡唑-5-甲酸50 g(0.16 mol),在搅拌下快速流加二氯亚砜150 g(1.26 mol)后继续常温搅拌5 min,然后升温至80℃,反应15 min后脱去二氯亚砜;继续向四口烧瓶中加入200 mL乙腈和2-氨基-5-氯-N,3-二甲基苯甲酰胺28 g(0.14 mol),缓慢升温至80℃,反应0.5 h后冷却至0-5℃,继续搅拌1 h后抽滤,滤饼用清水洗涤2-3次,于80~90℃烘干,得白色粉末状产品74.1 g,HPLC归一含量95.5%,收率94%,反应总收率51.09%(以2,3,6-三氯吡啶计)。
熔点:209.4℃ (文献值208-210℃),样品经质谱确认相对分子质量为483.1,与文献值一致。1H NMR(400 MHz,DMSO)δ:10.26(s, 1H,PhNH), 8.49(dd, 1H,Pyridin-6-H), 8.25(dd,1H,Pyridin-4-H),8.17,(dd,1H,Pyridin-5-H),7.62(d,1H,Ph-H),7.47(d,1H,Ph-H),7.39(s,1H,Pyrazole),7.34(d,1H,NH).2.66(s,3H,-NH-CH3), 2.16(s, 3H, Ph-CH3)。
将本文所开发的合成方法与杜邦公司所开发的以2,3-三氯吡啶为原料合成氯虫苯甲酰胺的方法(简称方法1)作对比,具体对比内容见表1,通过对比可以得出以下结论:
表1 合成方法对比

本文采用价格较为低廉的2,3,6-三氯吡啶替代2,3-二氯吡啶为原料,避免了2,3-二氯吡啶原料难得、价格昂贵等缺点;与方法1相比,利用溴化及水解反应一锅法减少了氧化反应步骤,降低了合成成本。
方法1采用甲基磺酰氯等管制类剧毒药品,反应危险性大且价格昂贵。而本文采用价格低廉的酰化试剂,既节约反应成本又降低了反应风险。
方法1中使用有机金属试剂并涉及2步低温反应,对设备要求较高,不易实现工业化生产。而本文以加氢还原、溴化水解一锅法等方式避免了低温反应,降低了对设备的严苛要求。
方法1采用过硫酸钾和浓硫酸氧化以及大量高沸点有机溶剂水解易产生大量废酸及同废,对环境有较大危害。而本文所述的合成方法中反应结束后的母液均可直接套用,减少了废液的产生,另外尾气大部分为氯化氢,氯化氢可作为副产直接回收利用,整个过程清洁环保,符合现代绿色化学要求。
本文以2,3,6-三氯吡啶为原料,经肼解、加氢还原、环合、溴代-水解得到3-溴-1-(3-氯-2-吡啶基)-4,5-二氢-1H-吡唑-5-甲酸,然后分别与SOCl2和2-氨基-5-氯-N,3-二甲基苯甲酰胺得到氯虫苯甲酰胺,避免了2,3-二氯吡啶原料难得、价格昂贵等缺点,降低了氯虫苯甲酰胺的合成成本;以价格低廉的酰化试剂代替甲基磺酰氯等管制类剧毒药品,提高反应工艺安全性;以母液多次循环套用减少固废和废水的产生,实现了环保生产。
综上所述,与现有氯虫苯甲酰胺合成方法相比,本方法具有反应条件温和、原料易得、操作简单、反应成本低、绿色环保等优点,具有工业化应用前景。